Content
How to use diArk's search modules 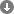
Information provided by diArk's result views
How to use diArk's search modules
The database is searched using modules that can be combined in chains. There are five different modules each providing specific options: a module for the full-text search in all species names, a taxonomy search module, a module to select specific groups of species, a module to search sequencing project related data, and a publication search module. A search can consist of any combination of modules and their options. By adding further search modules the user can successively refine the search and narrow down the result list. For each module the resulting selection of species, projects and publications is shown, providing additional context. When a new module is added the options available are restricted by the selection from the previous modules. At any time, the search options for every module can be changed and modifications are propagated down the chain reapplying previous user actions.
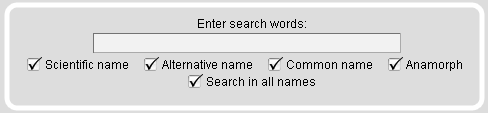
 Search in scientific and common names |
 Browse taxonomies and model organisms |
 Select specific species groups |
 Search by author, year or title |
 Search by sequencing project |
 Search by genome file |
Note: All species results will be listed under the reference scientific name (according to the NCBI taxonomy browser). Thus anamorphs will be listed under their teleomorph names (e.g. if you are searching for Fusarium verticillioides the main entry will be Gibberella moniliformis citing F. verticillioides as anamorph) and the same accounts for alternative scientific and common names.
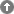
 Species Names Search Module
Species Names Search Module
The full-text search module provides an autocompletion input field to search the list of scientific and common species names. The default setting is that all different subsets of species names are marked. You can use your mouse or keyboard to select a species from the autocompletion list but you have to press "enter" a second time to submit your selection.
Note: If you just enter a single letter you will get the list of all species starting with this letter, depending on your selection of scientific, alternative and/or other names.
 Taxonomy Search Module
Taxonomy Search Module
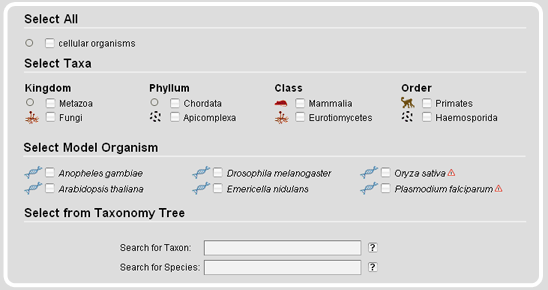
The Taxonomy Search Module offers tables containing specific subsets like a selection of major taxa or a range of model organisms. Several different strains have been sequenced for some of the model organisms. Those are marked with exclamation mark icons. In addition, on mouse-over the species names the complete scientific strain names are given.
Note: If the dataset has been restricted by previous modules (e.g. the selection of a specific reference), excluded species and taxa are disabled in the tables and will not be shown in the taxonomic tree representation.
The taxonomy search module also provides a taxonomic tree representation for the selection of taxa and species. On top of the tree representation, there are two autocompletion input fields, one for taxa and one for species names. The species name input field accepts all different types of names, the scientific names, the alternatively used names, common names, and the anamorphs, while it selects the scientific reference name. After submission of the taxon/species name search, the taxonmy tree is reloaded with the selected taxon/species name opened.
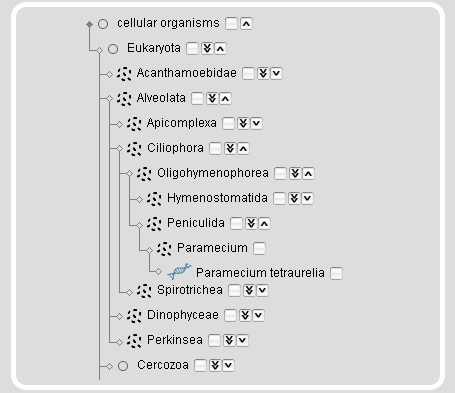
Instead of searching for taxa or species names, taxa and species can be browsed and selected by expanding/collapsing and including/excluding subsections of the tree.
| Clicking the arrow-down icon will expand the tree by 1 taxon. | |
| Clicking the double arrow-down icon will expand the tree by 5 nodes. On mouse-over the icon the number of sub-taxa and species will be given. | |
| Clicking the arrow-up icon will collapse the subtree. |
On mouse-over a species name an image of the species will be shown. Selecting a taxon will select the complete subtree and all species included. This will also select all taxa/species that are not shown based on restrictions in previous modules. The rationale is, that you might want to change your restrictions in a previous module but still want to keep your taxonomic selection. For example, if you restricted your search to genomic sequences in the projects module and selected the Arthropoda you will only see Arthropoda species with sequenced genomes. If you then change your selection in the projects module to also include the cDNA/EST projects, your species selection will automatically expand to include all Arthropoda for which genomic sequences and/or cDNA/EST data are available.
Note: Expanding the complete tree or major taxa (e.g. the Fungi/Metazoa group) will cause Firefox to take some time to render the updated view.
 Species Groups Search Module
Species Groups Search Module
With the Species Groups Search Module specific subsets of species can be selected that do not necessarily correspond to certain taxa. We plan to expand this tool to show more general information about the species groups listed. These informations would include for example the ratio for the selection of the four Plasmodium falciparum strains for genomic sequencing. At the moment, only subsets of species can be selected.

 Publication Search Module
Publication Search Module
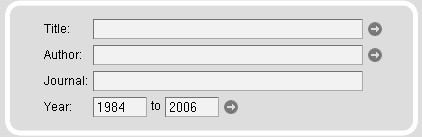
The publications related to the species and sequencing projects can be searched in several ways. Full-text searches are provided for titles, authors, abstracts, and journals. In addition, the journal search input field is supplemented with an autocompletion function. Searches can be restricted by dates. By default, searches are unrestricted.
Note: Only by selecting the Publications search module, the species and sequences are already restricted to those whose genome or cDNA/EST data or sequence has been referenced in a publication.
 Projects Search Module
Projects Search Module
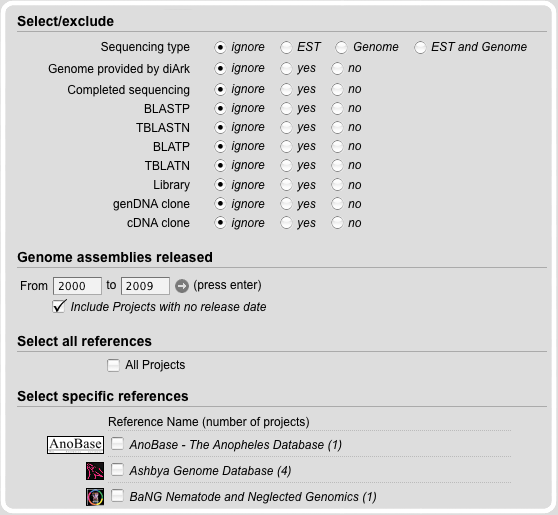
The projects contain details concerning a specific sequencing effort, such as its type (genomic DNA or EST/cDNA) and a link to the web-page providing the primary data. Projects can be selected based on the sequence data available for the species. Thus, species might be selected for which only cDNA/EST data or only genomic data is available, or those for which both cDNA/EST and genomic data is available. The term completeness is intended to describe the coverage of the genome and the chance to find all homologs of the gene of interest. For detailed information have a look at the FAQ's pages. The completeness option offers the possibility to search for those species for which the chance to find all homologs (and complete genes) to the protein of interest is very high. Projects can also be selected based on several services provided, like BLASTP or TBLATN.
The genome assemblies release option is a nice tool to select those genomes that have been finished in a certain time scale. Thus, the history of the genome sequencing efforts can be analysed (of course with limitations).
The projects have been organized by references, a term we use for the large-scale sequencing centers (e.g. the DOE Joint Genome Insitute) or community species homepages (e.g. FlyBase). By choosing references it is possible to search for the output of a single or more sequencing centers. However, for many species, the sequence information is not available via a dedicated species home page but only via GenBank. Therefore the "GenBank" links (associated to the GenBank reference) of the species projects provide BLAST search forms including the corresponding database (some data is only available from the WGS, other from the EST database) and the corresponding species name.
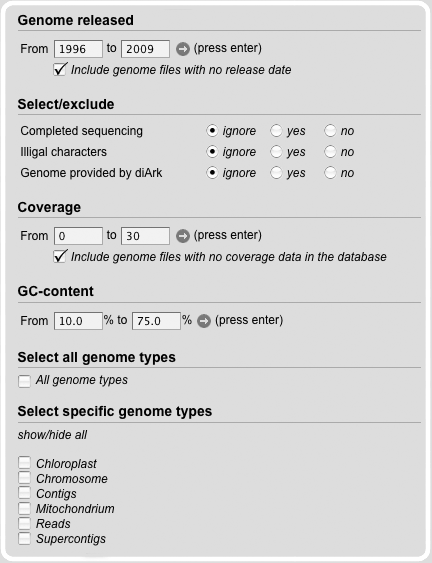
 Genome File Search Module
Genome File Search Module
The Genome File Search Module provides fields, to limit the search. Therefore, the release date of the genome file can be set.
Furthermore, information about completeness, illegal characters, the coverage, the GC-content and the genome type can be adduct.
The default setting is that all different subsets are marked
Information provided by diArk's result views
Species Result View
This view is the central gateway to the species data. Species can be sorted by either taxonomy or name. The species view provides the full taxonomy, alternative scientific names, common names, names of anamorphs, comments to species, the list of linked projects, and a list of publications.

Publications Result View
This view provides information on publications including authors, titels, journals, and abstracts.

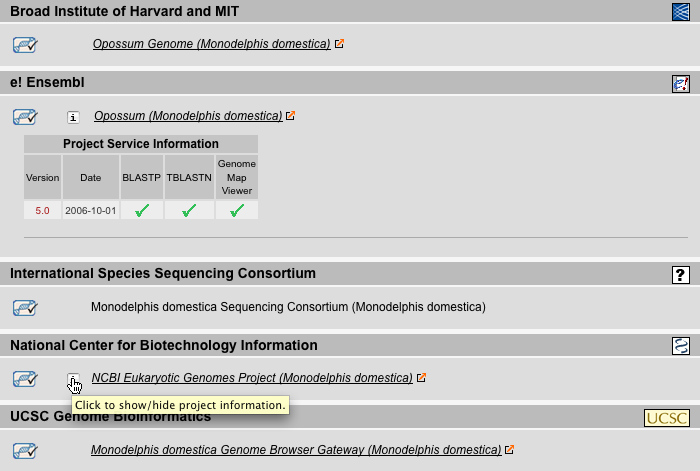
Projects Result View
The Projects Result View lists the sequencing projects sorted by sequencing centers (called references). For some projects additional information is available by clicking on the 'i'-icon. This includes BLAST- and BLAT-services provided, genome map viewer, and DNA-libraries.
Genomes Result View
The Genomes Result View lists the species sorted by either taxonomy or name. It provides a table with several genome related data: (A) The number of chromosomes (if known). (B) The size of the genome given as number of base pairs included in the chromosome-, supercontigs-, or contigs-file, in descending priority. This is just a rough estimate because the chromosome- and supercontigs-files in most cases do not contain the unplaced supercontigs and contigs, respectively, which reduces the number of base pairs, but instead contain undefined bases "N" either in variable numbers (estimated e.g. from BAC-clone lengths) or by always including 50 or 100 bp which increases the number of bases. (C) the GC content in percent. (D) The number of entries in the highest priority file (chromosome > supercontigs > contigs).
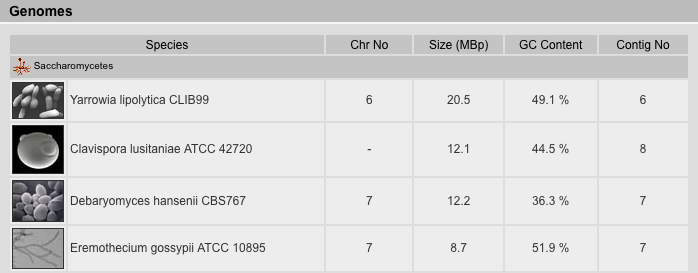
Genome Files Result View
The Genome Files Result View lists information corresponding to genome assembly files. The files are sorted by species followed by sequencing centers. The type of the file is inserted manually and can be one of the following:
| Chromosome | Every fasta-entry in this file is a chromosome. There are a few exceptions where chromosomes are split in two or more fasta-entries. |
| Uchromosome | These files contain contigs/supercontigs which could not be mapped to any (unknown chr.) or anchored (random chr.) to a certain chromosome. |
| Supercontigs | Every fasta-entry represents a supercontig which consist of sorted contigs separated by estimated or fixed numbers of "N" bases. |
| Contigs | Contigs (from "contiguous sequence") are the smallest pieces of an assembly and consist of overlapping sequence reads. |
| Ureads | These files contain the unplaced reads, reads that could not be assembled to contigs. These files are especially important for low-coverage genomes that in most cases end up with very short contigs. In these cases, small proteins or some exons can be reconstructed from the ureads-files. |
| Apicoplast | These files contain the apicoplast DNA. The apicoplast is a relict, non-photosynthetic plastid found in Apicomplexa. It is proposed that it evolved via secondary endosymbiosis. The apicoplast is surrounded by four membranes within the outermost part of the endomembrane system. |
| Chloroplast | These files contain the chloroplast DNA. Chloroplasts are organelles found in plant cells and eukaryotic algae that conduct photosynthesis. |
| Kinetoplast | These files contain the kinetoplast DNA. A Kinetoplast is a disk-shaped mass of circular DNAs inside a large mitochondrion that contains many copies of the mitochondrial genome. Kinetoplasts are only found in protozoa of the class kinetoplastea. Kinetoplasts are usually adjacent to the organisms' flagellar basal body leading to the thought that they are tightly bound to the cytoskeleton. |
| Mito | These files contain the mitochondrial DNA. Mitochondria are membrane-enclosed organelles found in most eukaryotic cells. |
In addition, there are other rarely used file types: Ultracontigs (very long supercontigs, but not mapped to chromosomes yet), Usupercontigs (contigs that could not be ordered to supercontigs).
Where available, we provide the version of the assembly as well as the release date of the data. In general, we have taken the versions and release dates as given by the sequencing centers. If those are not provides, we have taken the dates on which the files were saved in the ftp-directories. For NCBI-assembly data, we have taken the dates on which the data has been submitted to NCBI. Note: Version numbers do not correlate between sequencing centers and NCBI! Assembly version 6.0 at a sequencing center might correspond to version 1.0 at NCBI because it was the first version submitted.
The completeness is the same as given in the projects view, and is a rough estimate of the completeness and quality of the data and assembly. In general, assemblies with coverages below 4 are regarded as incomplete.
The genome coverage of the assembled sequence data is given if it is provided by the sequencing centers.
The GC content, the size in Giga-base-pairs, the number of fasta-entries ("contigs"), the occurrence of illegal characters in the sequences (not beeing g/G, a/A, t/T, c/C, or n/N), and the typical length of the fasta-entries were calculated from the fasta files.
For genome assemblies available from NCBI, the accession numbers can be shown by clicking on "Acc." and the assemblies are provided as zipped fasta files.
For some assemblies, comments are available that provide further background information about differences to earlier assemblies, problems in the assembly process, and others.
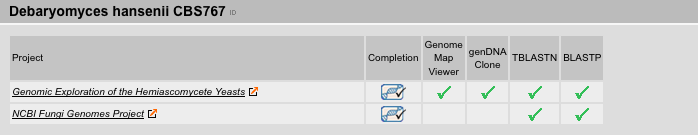
References Result View
The References Result View lists the sequencing projects sorted by species. Links to the sequencing centers, the status of the sequencing project (complete or incomplete, sequencing of genomic DNA or cDNA/EST), as well as additional information including the availability of BLAST- and BLAT-services, genome map viewer, and DNA-libraries are provides. Here, the user can directly access the sequencing project webpage most suitable for his purposes.
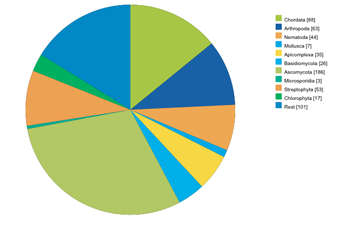
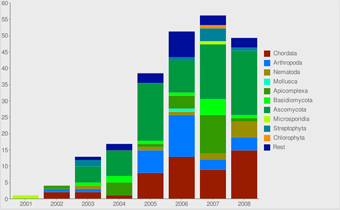
Sequencing Statistics View
Here, some statistics regarding the sequencing efforts are presented. Graphs are related to all projects, EST/cDNA sequencing projects and whole-genome-sequencing projects. Also, some graphs are shown plotting the assembled genomes against the time of the release of the data.